Main Program/Method name: dnaPipeTE
Main Program/Method version (if applicable): 1.4c ("container")
Tutorial Author(s): Clément Goubert
Last Update: 02-23-2023
¶ Description
dnaPipeTE (for de-novo assembly & annotation Pipeline for Transposable Elements), is a pipeline designed to find, classify and quantify Transposable Elements and other repeats in low coverage (< 1X) NGS datasets. It is very useful to quantify the proportion of TEs in newly sequenced genomes since it does not require genome assembly and works directly on raw short-reads.
¶ Objectives
- Install dnaPipeTE on local machine or server via Docker or Singularity
- Execute a complete interactive run of dnaPipeTE using an example dataset (Drosophila melanogaster)
- Process outputs to obtain summary graphs (TE content, TE landscapes)
¶ Prerequisites
¶ hardware
- OS: Linux or MacOS X
- CPU: 1 (8 or more recommended)
- RAM: 10Gb (16G recommended)
- Storage: <10Gb
¶ software
- Docker (MacOS or Linux with root privileges)
- Singularity (Linux machines without root privileges)
¶ Protocol
¶ 1. Installation
Pull and test the dnaPipeTE container according to system and distribution (chose the tab corresponding to your need).
docker run -it clemgoub/dnapipete:latest
# within the container (the prompt will be "(dnaPipeTE/opt/dnaPipeTE)#")
python3 dnaPipeTE.py -h # this will display the help screen
exit # return to the host machine shell
# first download the image on your machine
mkdir ~/dnaPipeTE
cd ~/dnaPipeTE
singularity pull --name dnapipete.img docker://clemgoub/dnapipete:latest
# then open an interactive shell
singularity shell ~/dnaPipeTE/dnapipete.img
# within the container (the prompt will be "<Singularity>")
cd /opt/dnaPipeTE
python3 dnaPipeTE.py -h # this will display the help screen
exit # return to the host machine shell
¶ 2. Data
- Raw short reads
In this tutorial, we will use the raw WGS 2x150bp Illumina reads generated by Lawlor et al., 2021 for female individuals of the line w1118. The original DNA has been extracted from 20 whole adults and homogenized before sequencing. dnaPipeTE only requires one end of paired-end data, and it can be either R1 or R2.

Idealy, short read data should be cleaned from non-nuclear host DNA. At the very least, mtDNA should be removed by aligning the raw reads to the mithocondrial genome of the target species.
For this tutorial, we have prepared a toy dataset that has already been filtered using bwa mem.
If no mithochondrial genome available for your species of interest, dnaPipeTE can be run on the raw read set to assemble the mtDNA genome. It can be easily recovered in the outputs as one of the largest (in bp) and most abundant repeat (see post-processing section)
- TE library
dnaPipeTE rely on a user-provided TE library in fasta format for the classification of the repeats. For best results, the header of the library should be formated in the "RepeatMasker" format >TEname#type/subtype.
A drosophila-specific TE library in the excpected format has been created for this tutorial.
Formatted libraries can be extracted from popular databases:
From DFAM, this can be done using the tool FamDB and the command:
famdb.py -i <Dfam.h5> families -f fasta_name -a <taxa> --include-class-in-name > library.fasta
- We have prepared a repository with ready-to-go input data as well as a folder the expected results. To set-up yout working environment with the required input, type the following commands in your terminal:
mkdir ~/Project
cd ~/Project
# download and extract the dataset
wget https://sandbox.zenodo.org/record/1122020/files/dnaPipeTE_Demo_Droso.tar.gz
tar -zxvf dnaPipeTE_Demo_Droso.tar.gz
# remove the archive
rm dnaPipeTE_Demo_Droso.tar.gz
The data structure should look like this:
# ~/Project
# └── data
# ├── Droso_TE.fasta
# └── SRR14470610.mt.clean.R1_2X.fastq
# └── dnaPipeTE_tutorial_results
¶ 3. Running dnaPipeTE
Now that the reads and TE library are downloaded in ~Project/data we can run dnaPipeTE!
¶ 3.1. Start the container
The first step is to launch an interactive session and make our ~Project directory accessible from within the container. The command is slightly different between Docker and Singularity, but the principle is the same. We will instruct the container where our files are on the physical machine and where we want to see them on the container:
docker run -it -v ~/Project:/mnt clemgoub/dnapipete:latest
singularity shell --bind ~Project:/mnt ~/dnaPipeTE/dnapipete.img
# within the container:
cd /opt/dnaPipeTE
¶
in both cases, the content of
~/Projectwill be seen as~/mntby the container. Thus~/Project/datawill be accessed with~/mnt/data
¶ 3.2. Run dnaPipeTE
The dnaPipeTE script must be executed in the source folder for the script, located in /opt/dnaPipeTE. By default, the container working directory is already set there, so there is nothing else to do but run dnaPipeTE!
For this tutorial, we will run dnaPipeTE with the following parameters:
- Trinity iterations (
-sample_number): 2 - Coverage per iteration (
-genome_coverage): 0.15X - RepeatMasker annotation threshold (
RM_t): 0.25 (25% of the query repeat must be annotated to keep a hit; lower this value for increased sensitivity)
python3 dnaPipeTE.py \
-input /mnt/data/SRR14470610.mt.clean.R1_2X.fastq \
-output /mnt/dnaPipeTE_0.15_1_t25 \
-genome_size 175000000 \
-genome_coverage 0.15 \
-sample_number 2 \
-RM_lib /mnt/data/Droso_TE.fasta \
-RM_t 0.25 \
-cpu 8
For this tutorial, we will run dnaPipeTE with the following parameters:
- Trinity iterations (
-sample_number): 2 - Coverage per iteration (
-genome_coverage): 0.05X - RepeatMasker annotation threshold (
RM_t): 0.25 (25% of the query repeat must be annotated to keep a hit)
python3 dnaPipeTE.py \
-input /mnt/data/SRR14470610.mt.clean.R1_2X.fastq \
-output /mnt/dnaPipeTE_mock \
-genome_size 175000000 \
-genome_coverage 0.05 \
-sample_number 2 \
-RM_lib /mnt/data/Droso_TE.fasta \
-RM_t 0.25 \
-cpu 8
To speed-up computations, we will only use samples of 0.05X. We will later explore the outputs and perform some post-processing using a realistic output generated with samples of 0.15X (
~/Project/dnaPipeTE_tutorial_resultsa.k.a/mnt/dnaPipeTE_tutorial_resultsin the Docker container)
¶
While the program runs, we can look in details what is happening:
- Our original read dataset
SRR14470610.mt.clean.R1.fastqis first sampled at 0.15X and fed toTrinitywhich will produce a first assembly of the repeats - Next, the reads that contributed to the assembly are kept and merged to a second fresh sample of 0.15X drawn from the input file. Thus, the second iteration of Trinity runs with a reads set <0.30X. We could continue these iterations as needed, but 2 is often a good starting point.
- Following the two iterations of
Tritnity, the final repeat assembly is produced (Trinity.fasta). - To identify known TEs among the assembled repeats,
dnaPipeTErunsRepeatMaskerusing the user-provided library (Droso_TE.fasta). - To quantify each assembled repeat in
Trinity.fasta, a new read sample of 0.15X is drawn fromSRR14470610.mt.clean.R1.fastqand mapped to the assembly (Trinity.fasta). The counts (inbpandreads) for each assembled contig are tabulated inreads_per_components_and_annotation.
¶ 4. Post-processing and data interpretation
Since its first version, dnaPipeTE produces a large (too large) amount of outputs. Several analyses included in dnaPipeTE turned out to be redudant, overly complicated and sometimes confusing. In order to sort out the wheat from the shaff, we will indicate here the most important outputs to consider, and generate new, more informative output graphs using the companion package dnaPT_utils.
¶ 4.1 Useful output files
Trinity.fasta:
This is the final assembly of the repeats. Each contig is named according to Trinity's nomenclature, which expect a hierarchical structure of RNA molecules: "read cluster" > "gene" > "transcript"; for example comp_TRINITY_DN374_c0_g1_i3. In the context of low-coverage DNA sequencing, we can infer a similar hierarchy for the assembled repeat sequences:
| Trinity header segment | RNA-seq | dnaPipeTE |
|---|---|---|
| comp_TRINITY_DN374_c0 | reads cluster | reads cluster |
| g1 | gene | suggest same superfamily/family |
| i3 | transcript/isoform | suggest same family, structural variants, related fragments |
Note that in the context of RNA-seq, the outputs of Trinity are expected to be fully-assembled transcripts. In dnaPipeTE, the output of Trinity are expected to be partially-assembled repeats, meaning that most contig produced will represent only a fragment of an actual family. Fully assembled TEs can occur if there is enough repeats in the genome and the sample coverage is high enough. However, it is not required for quantifications.
read_per_component and annotation:
This file is certainly the most important of all outputs. It includes the counts in bp and reads for each repeat contig present in Trinity.fasta, and report the annotation passing the threshold -RM_t. Note that contigs with no read mapped are not shown. This can happen when a low-frequency repeat is assembled, but no read to support it are present in the quantification sample.
head /mnt/dnaPipeTE_tutorial_results/reads_per_component_and_annotation
ouput:
2964 440760 comp_TRINITY_DN374_c0_g1_i2 10583 R2_DM LINE/R2 0.34064065009921574
1257 184441 comp_TRINITY_DN390_c1_g1_i1 7909 ROO_I LTR/Pao 0.9519534707295486
1150 169137 comp_TRINITY_DN405_c9_g1_i1 2390 R1_DM LINE/R1 0.999581589958159
944 138783 comp_TRINITY_DN405_c9_g2_i1 2930 R1_DM LINE/R1 0.8
711 105121 comp_TRINITY_DN342_c0_g1_i16 258 SAR_DM Satellite 0.9961240310077519
700 103785 comp_TRINITY_DN379_c4_g1_i3 264 SAR_DM Satellite 0.9962121212121212
595 88236 comp_TRINITY_DN387_c2_g1_i1
| reads | bp | contig | contig length | RM hit name | RM hit class | annotation prop. |
|---|---|---|---|---|---|---|
| 2964 | 440760 | comp_TRINITY_DN374_c0_g1_i2 | 10583 | R2_DM | LINE/R2 | 0.34064065009921574 |
| 1257 | 184441 | comp_TRINITY_DN390_c1_g1_i1 | 7909 | ROO_I | LINE/R1 | 0.9519534707295486 |
- More details about the dnaPipeTE outputs can be found here.
¶ 4.2 Generating summary graphs with dnaPT_utils
dnaPT_utils is a companion repository with a few post-processing script. The complete documentation with examples can be found here
- TE quantification
dnaPT_charts.sh -I /mnt/dnaPipeTE_0.15_1_t25 -o DM
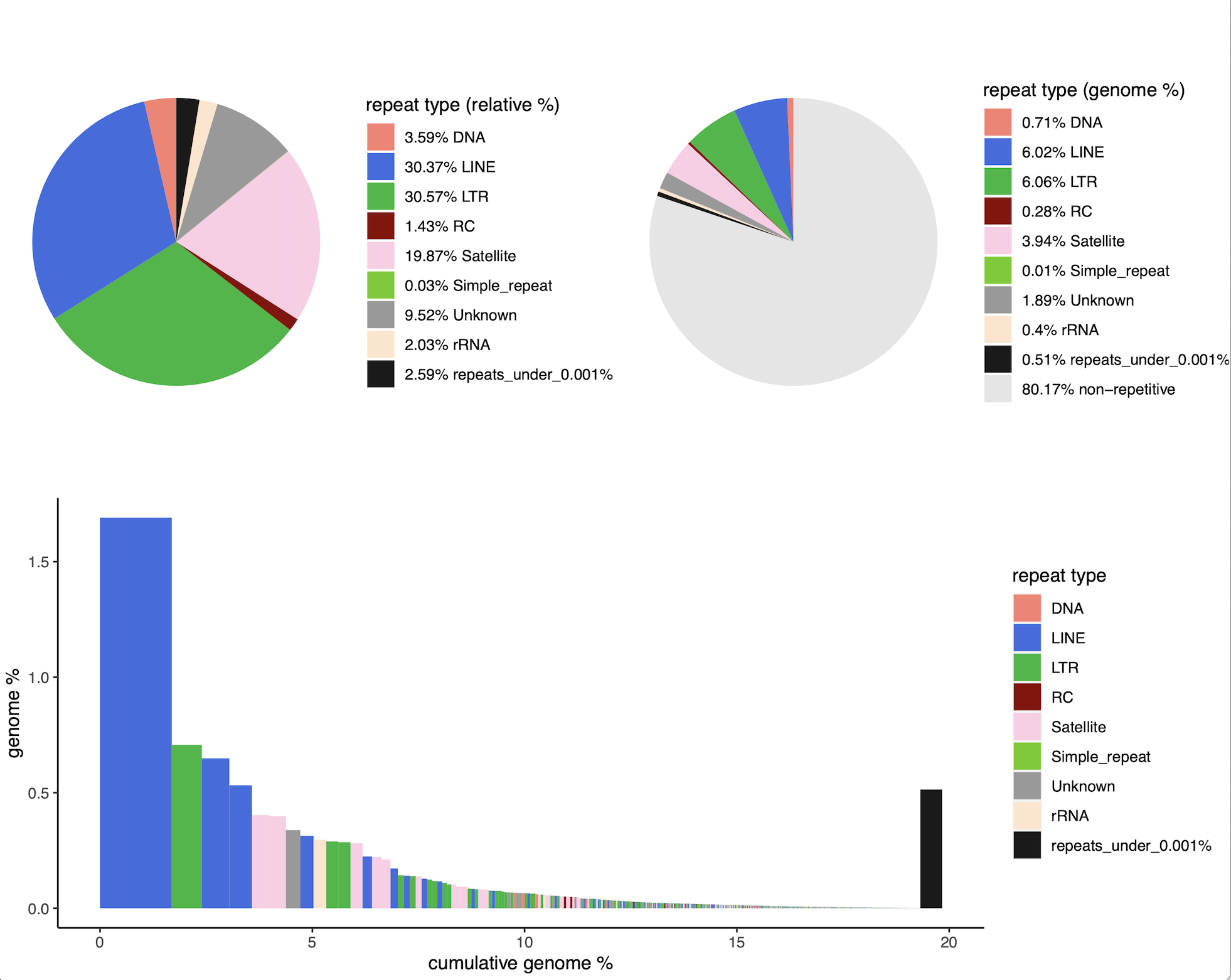
- TE landscapes
dnaPT_landscapes.sh -I /mnt/dnaPipeTE_0.15_1_t25 -p DM
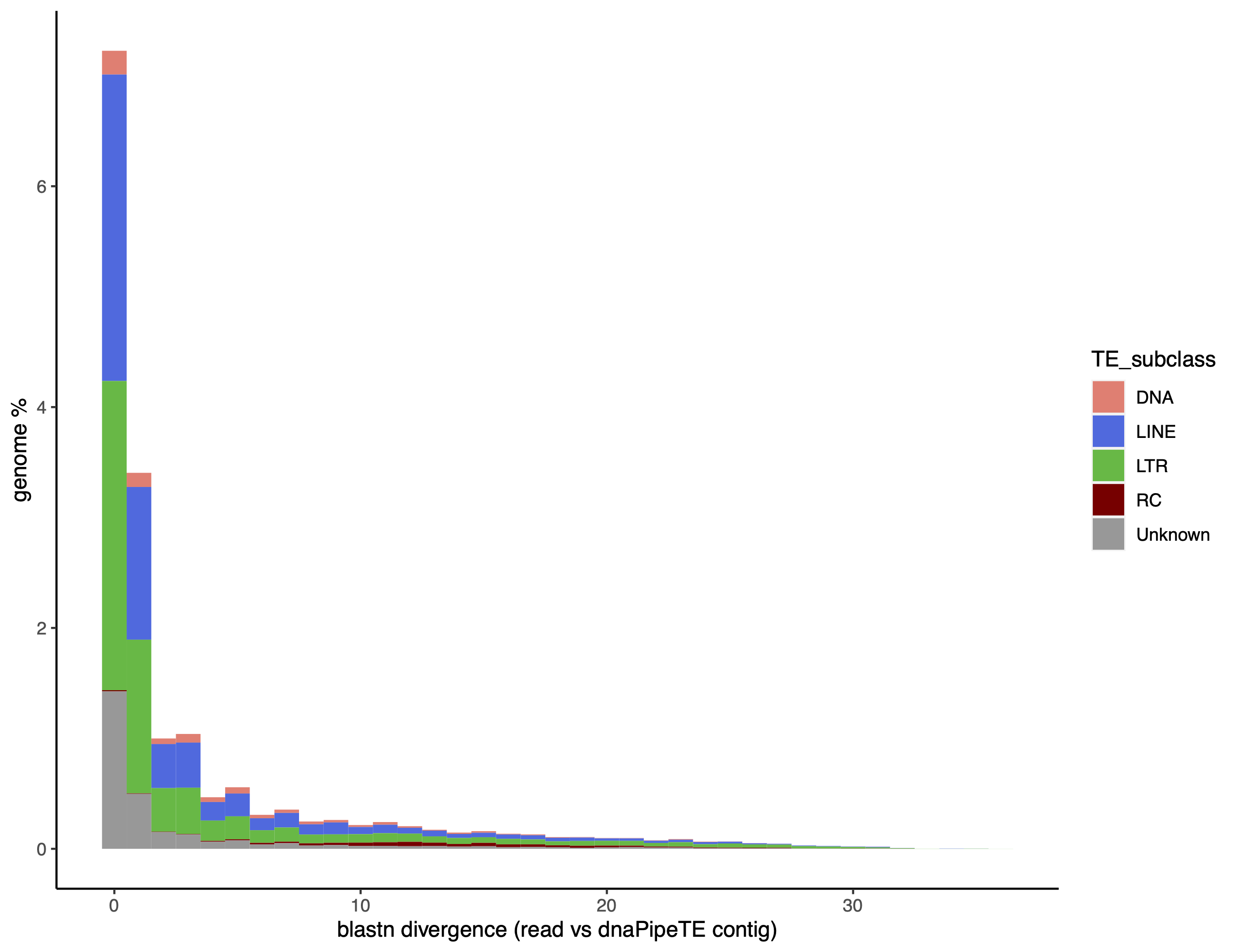
For landscapes at the superfamily level -S:
dnaPT_landscapes.sh -I /mnt/dnaPipeTE_0.15_1_t20 -p DM -S
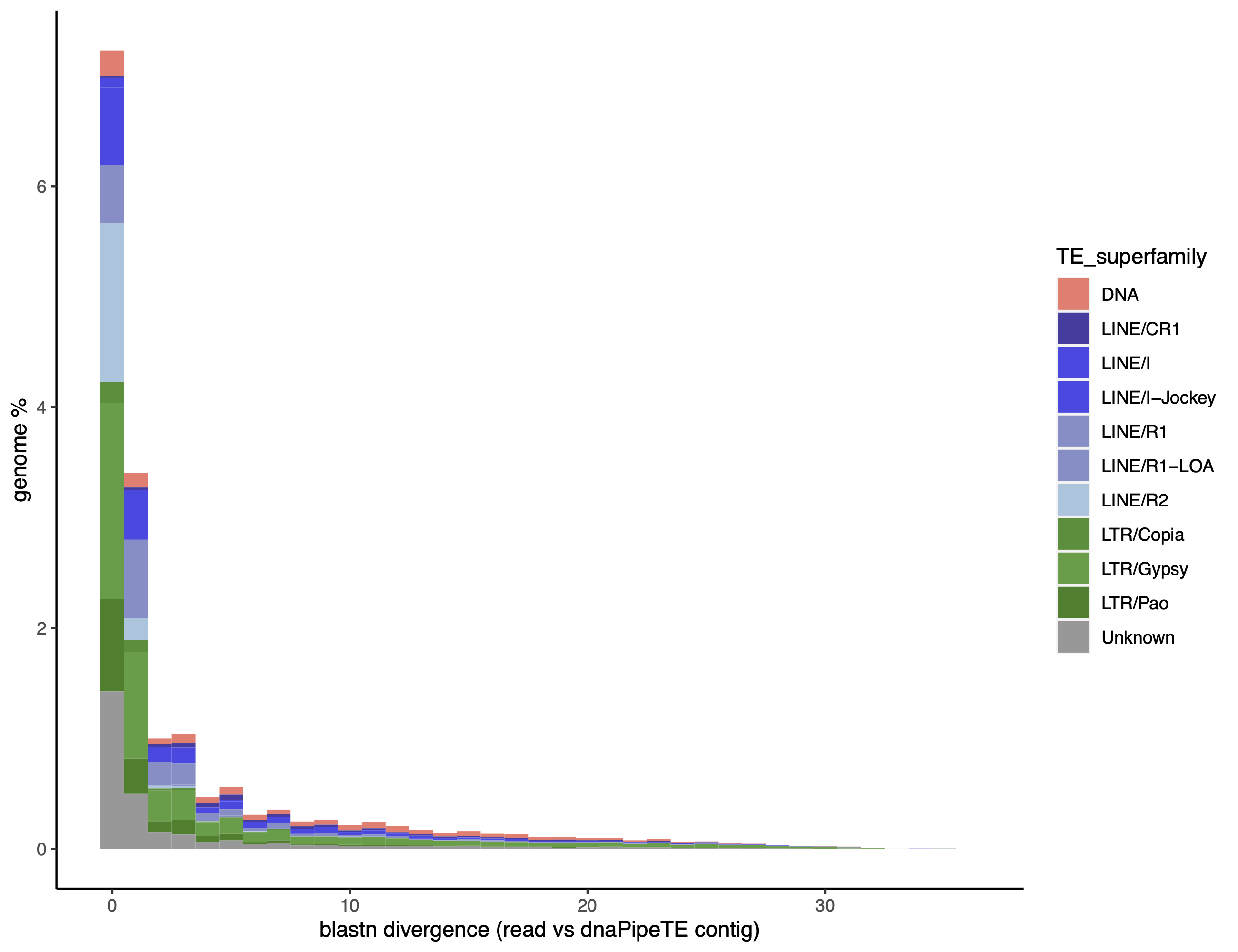
For landscapes at the superfamily level -S:
dnaPT_charts.sh -I /mnt/dnaPipeTE_tutorial_results -o DM
- TE landscapes
dnaPT_landscapes.sh -I /mnt/dnaPipeTE_tutorial_results -p DM
dnaPT_landscapes.sh -I /mnt/dnaPipeTE_tutorial_results -p DM -S
- Download the graph on your local machine. Open a new terminal window/tab and type:
sftp <user>@<IP>:/home/<user>/Project
get dnaPipeTE_tutorial_results/DM_*
exit